



Organic electronics have advanced rapidly since the demonstration of the first bi-layer organic light emitting device in 1987 [1], with OLEDs now ubiquitous in high-end mobile phones, smart devices, and consumer TVs. Although organic solar cells (or organic photovoltaics, OPVs) are yet to see the same commercial success as OLEDs, at one point appearing as if new material systems such as perovskites may disrupt them entirely, a recent resurgence in material design and device performance has brought them firmly back into attention as a viable clean-energy solution. Kurt J. Lesker has been providing vacuum deposition systems dedicated to the production of organic electronic devices for over 20 years, with many systems globally in university and R&D laboratories.
Although the power conversion efficiency (the ability of the solar cell to convert light energy into useful electrical energy) is often the headline-grabbing parameter, device stability is equally important if the technology is to become a commercially viable, long-lasting product. There are many possible degradation pathways in an OPV device, particularly unwanted chemical reactions with atmospheric contaminants (e.g. oxygen and moisture) and chemical, morphological or interfacial changes caused by mechanical, illumination, thermal and electrical stresses during operation. Understanding the origins of the degradation pathways is critical to determine what steps can be taken to mitigate them to allow stable device performance. For example, a material which undergoes photo-oxidation may be protected through encapsulation to prevent the ingress of oxygen, whilst that is unlikely to help a film which suffers damaging morphology changes under thermal stress.
One particularly important degradation pathway is the photo-dimerization of fullerene materials, initially reported in 1993 by Ecklund et. al. [2], due to the heavy use of fullerene-based acceptor materials in OPVs and the fact that it appears resistant to standard encapsulation techniques. Photo-dimerization in C60 has been found to reduce the exciton lifetime and diffusion length, resulting in a decrease in current generation of the solar cell device [3]. Early investigations suggested that the reaction proceeds by a [2+2] cycloaddition and is initiated by the presence of T1 triplet excitons in the fullerene [2,4]. In a recent publication in Nature Communications titled “The role of spin in the degradation of organic photovoltaics” (https://doi.org/10.1038/s41467-020-20601-6) [5], an international collaborative team (Heliatek GmbH, University of Oxford, KAUST, TU Dresden, Julius Maximilian University of Würzburg, Hasselt University), with lead authors Ivan Ramirez (Heliatek, GmbH) and Moritz Riede (University of Oxford), set out to understand the physical pathway which results in this dimerization reaction, with particular focus on the formation of the fullerene triplet excitons.
To observe the dimerization reaction, the team produced vacuum deposited solar cells and related thin film structures based on dilute electron donor:C60 co-deposited blends using various electron donor materials, with the complete solar cells fabricated in a Kurt J. Lesker Spectros evaporation system. The donor materials were selected to give a wide range of energetic and electronic properties, allowing the critical device parameters to be determined, whilst (mostly) absorbing little light themselves so that the impact on the C60 could be isolated through optical spectroscopy techniques. In the initial experiments, UV-visible optical absorption spectra and external quantum efficiency scans (EQE, the monochromatic efficiency of the solar cell as a function of wavelength) were recorded over a period of time for the various device structures. The team uncovered some interesting results; some blends (e.g. TAPC:C60) resulted in significant changes to the absorption spectra and EQE, particularly around 320nm and 395nm which are indicative of C60 dimerization, whilst others (e.g. m-MTDATA:C60) resulted in no significant changes at these points. Although the electron donor material was only present in a dilute quantity (~5%), it appeared to be critically important to the dimerization reaction.
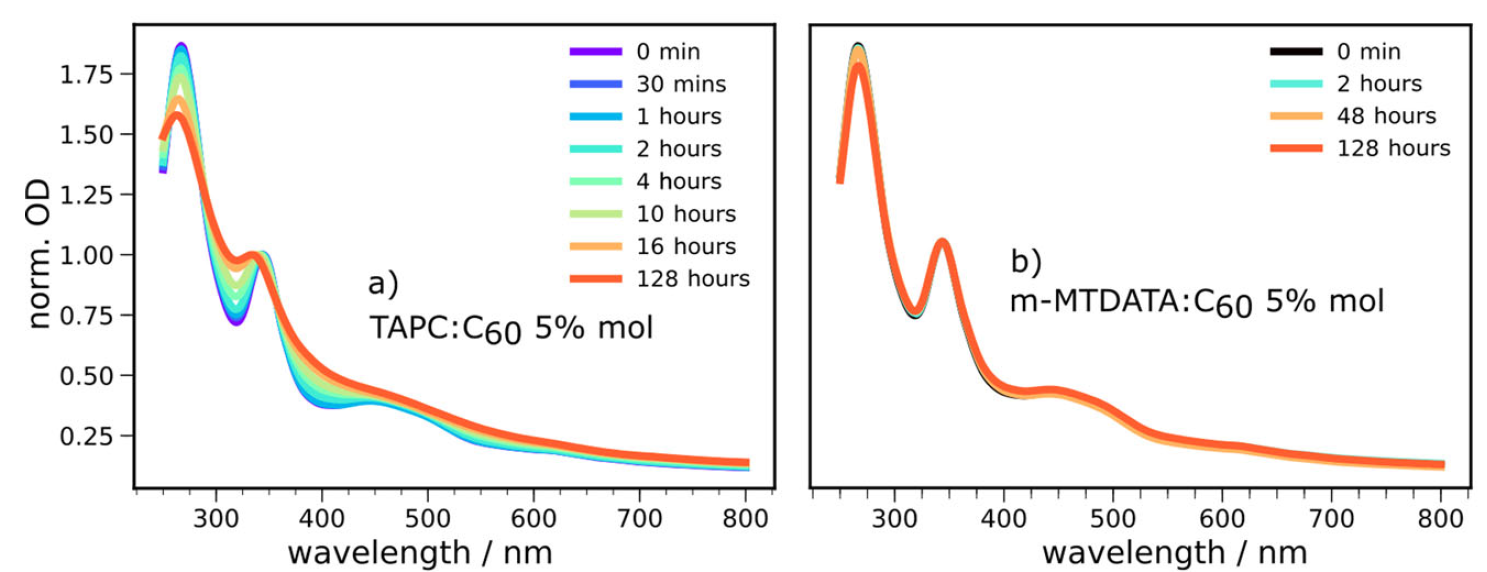
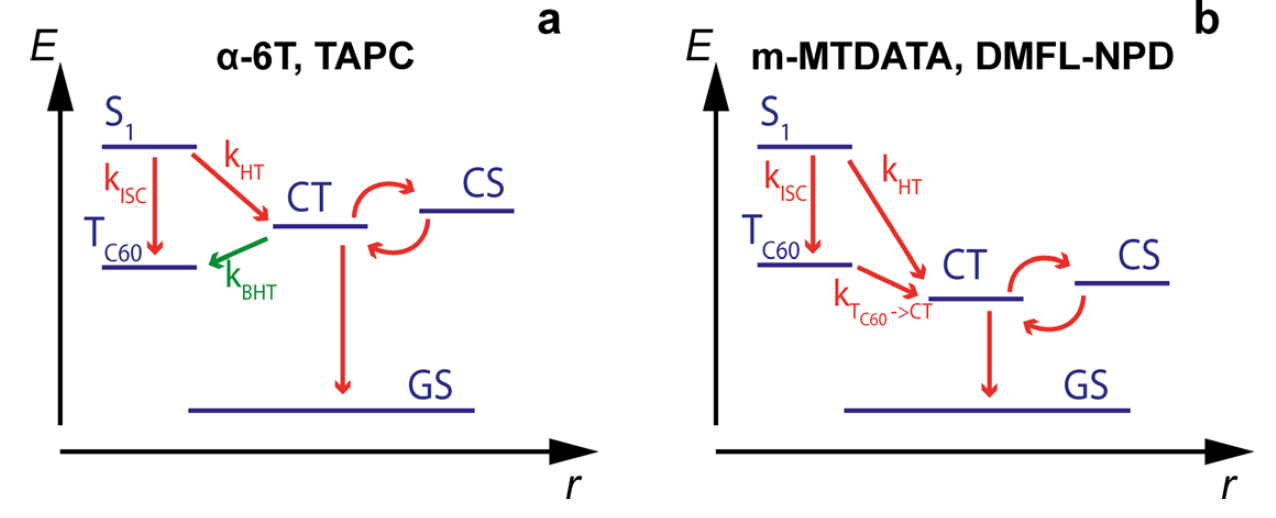
To understand the role of the electron donor material, the team considered the energetic pathways which could lead to the generation of the C60 triplet excitons (TC60) believed to be responsible for the dimerization reaction. Typically, triplet excitons would be generated by a process called intersystem crossing (ISC) where a singlet exciton (S1), formed when the material absorbs a photon of light, is converted directly into the triplet exciton. However, in the presence of an efficient electron donor material these singlet excitons are rapidly quenched via hole transfer (HT) into a charge-transfer state (CT - the precursor to the complete charge separation state, CS, required for the device to produce useful free charges) and should result in a significant reduction in ISC. The fact that dimerization is equally prominent in the presence of an efficient electron donor material as in a pure C60 film suggests ISC is not the only pathway to the triplet excitons. Instead, the team identified another potential route to these triplet excitons, a process called back-hole transfer (BHT), whereby an exciton which has already undergone hole transfer to the CT state is able to recombine back to the triplet exciton state through hole transfer back to the C60. Critically, the energy of the CT state dictates whether this pathway is viable; if the CT state is higher in energy than the TC60 state this BHT pathway is energetically favourable and is likely to contribute to a triplet exciton population, whilst if the CT state is lower in energy than the TC60 state the opposite is true, not only is BHT not possible, but the quenching of any TC60 excitons formed by ISC to the CT state becomes possible. Comparing this understanding to their earlier absorption and EQE experiments, the team identified that the electron donor materials which provided a CT level below the TC60 level (such as m-MTDATA) showed no indication of C60 dimerization, whilst those which provided a CT level above the TC60 level (such as TAPC) did exhibit the changes associated with dimerization. They were therefore able to confidently assign the pathway to the dimerization reaction as being the presence of BHT from the CT state.
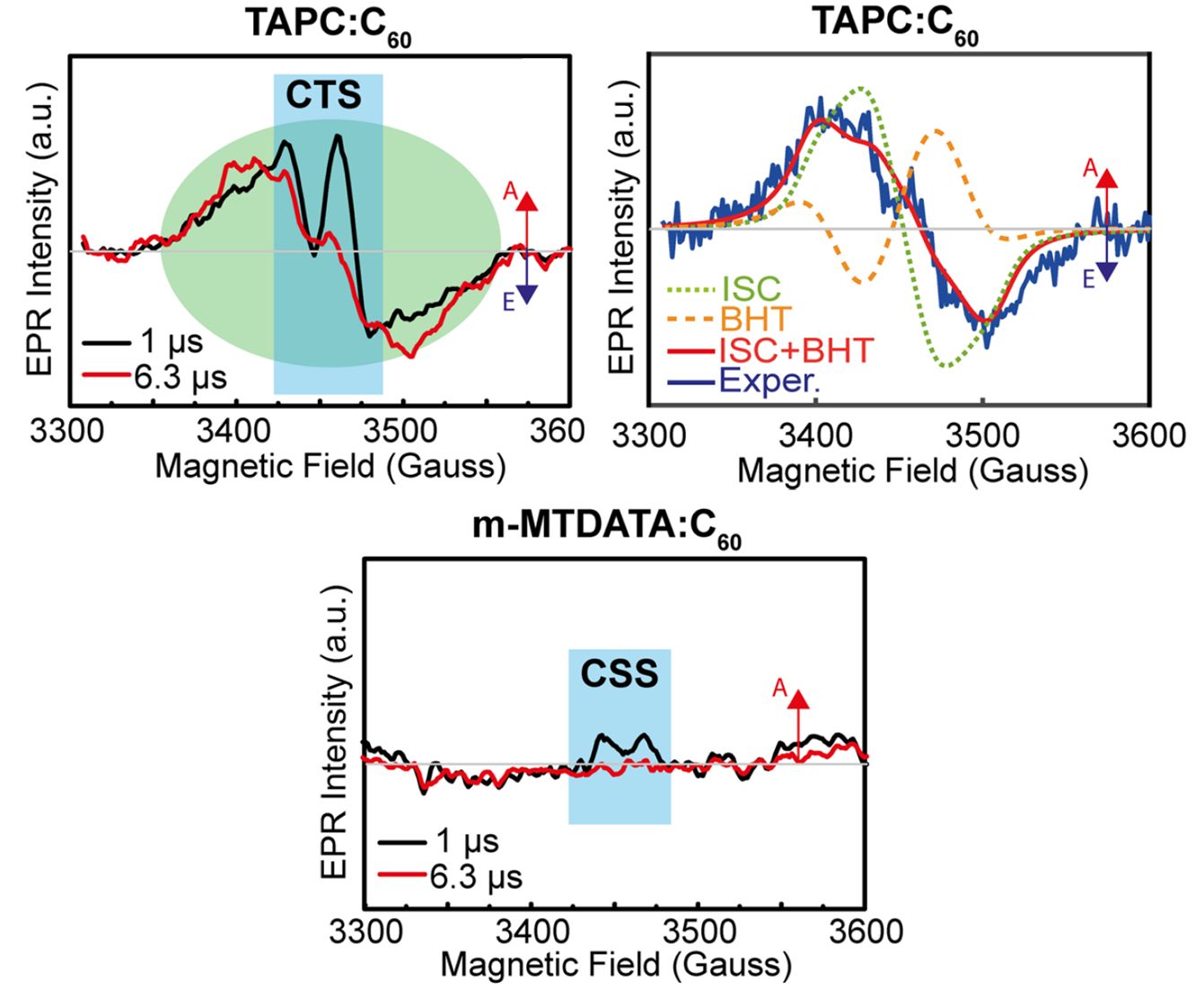
To confirm the triplet formation pathway, time-resolved electron paramagnetic resonance (TREPR) was used to observe the presence and origin of the C60 triplet states. For donor materials which provide a CT state at higher energy than TC60 (where BHT may be expected, e.g. TAPC) a significant spectral signal is seen. At a delay of 1 μs after the laser pulse, the spectra show a sharp signal (attributable to the CT state) superimposed on a broader background signal. After 6.3 μs the sharp CT signal has disappeared, leaving only the broad background signal which is caused by the C60 triplet excitons. This broad background signal was further analysed to identify the origins of the triplet state, with significant contributions seen from both ISC and BHT. As the TREPR measurements were performed at cryogenic temperatures (80K) the kinetics of the system will be significantly slowed compared to a device operating at room temperature and this may result in an enhancement of the ISC signal, though the presence of the BHT signal is clearly indicative of this as a viable pathway. For donor materials which provide a CT state at lower energy than TC60 (where BHT may not be expected, e.g. m-MTDATA) no significant TREPR signal is seen. This suggests suppression of triplets generated both via BHT (due to the prohibitive energy offset), and also ISC (as any ISC is likely followed rapidly by transfer to the CT state).
Whilst the result is important for the understanding of the degradation mechanisms in OPV devices, it raises some concerns over future device designs. As the viability of the BHT pathway is dependent only on the energetic operation of the device it cannot be mitigated through environmental controls, encapsulation or even improvements in exciton quenching. Careful material design is likely to be required to prevent this degradation mechanism and will be an important consideration for future high-efficiency OPV materials.
The work was supported, amongst others, by the European Union's Horizon 2020 research and innovation program under Marie Sklodowska Curie Grant agreement number 722651 (SEPOMO) and STFC Challenge Led Applied Systems Programme (CLASP, Grant number ST/L006294/1), both of which Kurt J. Lesker were proud to be involved with as industrial partners.
The original publication, including the figures reproduced in this report, was licensed under a Creative Commons Attribution 4.0 International Licence (http://creativecommons.org/licenses/by/4.0/) which permits reproduction in any format provided appropriate credit is given to the original authors. The figures are copyright © the article authors.
References:
[1] Tang, C. W. & Vanslyke, S. A., Organic electroluminescent diodes, Appl. Phys. Lett., 51 (12): 913 (1987).
[2] Rao, A. M. et al., Photoinduced polymerization of solid C60 films, Science, 259, 955–957 (1993).
[3] Burlingame, Q. et al., Photochemical origins of burn-in degradation in small molecular weight organic photovoltaic cells, Energy Environ. Sci, 8, 1005–1010 (2015).
[4] Wang, Y., Holden, J. M., Dong, Z. H., Bi, X. X. & Eklund, P. C., Photodimerization kinetics in solid C60 films., Chem. Phys. Lett., 211, 341–345 (1993).
[5] Ramirez, I., Privitera, A., Karuthedath, S., Jungbluth, A, Benduhn, J, Sperlich, A, Spoltore, D, Vandewal, K, Laquai, F & Riede, M., The role of spin in the degradation of organic photovoltaics, Nature Comm., 12, 471 (2021) (https://doi.org/10.1038/s41467-020-20601-6)